By Brian Kuzdas | Enterprise IT Architect | Madison, WI LinkedIn: https://linkedin.com/in/brian-kuzdas-a176b945
Picture this: your company just signed a contract with a mid-sized pharmaceutical manufacturer to implement a new cloud-based Manufacturing Execution System (MES). Six months in, a regulatory consultant walks through the implementation and says four words that stop the project cold: “Have you validated this?” Not for functionality. Not for performance. For FDA 21 CFR Part 11 compliance.
If you’ve worked in life sciences, biopharma, medical devices, or clinical research — even tangentially — you’ve encountered the shadow of FDA 21 CFR Part 11. And if you haven’t encountered it yet but work in any of those adjacent industries, you will. The regulation governs electronic records and electronic signatures used in FDA-regulated environments, and its reach is far broader than most IT architects initially expect.
This guide is written for enterprise IT architects, solution architects, cloud engineers, and technology leaders who need to understand what Part 11 actually requires, where compliance breaks down, and how to architect systems that will survive an FDA audit. I’ve spent 25 years building and validating systems in regulated environments. What follows is the practical knowledge I wish I’d had at the beginning.
1. What Part 11 Actually Requires
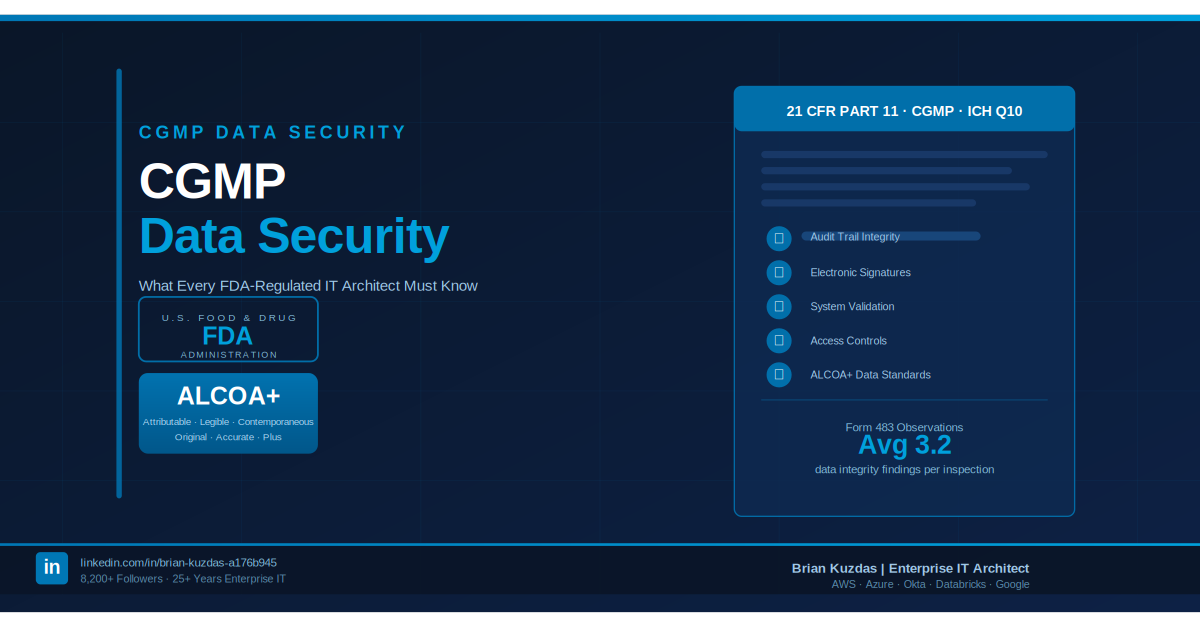
Title 21 Code of Federal Regulations Part 11 (21 CFR §11.1–11.300) establishes the criteria under which the FDA will consider electronic records and electronic signatures to be trustworthy, reliable, and equivalent to paper records and handwritten signatures. Published in final form in 1997 — years before modern cloud platforms, SaaS applications, or mobile computing existed — it remains the foundational compliance framework for any FDA-regulated industry using electronic systems.
Electronic Records (§11.10): Closed systems used to create, modify, maintain, archive, retrieve, or transmit electronic records must employ procedures and controls including:
- System validation: Systems must be validated to ensure accuracy, reliability, consistent intended performance, and the ability to discern invalid or altered records.
- Audit trails: Secure, computer-generated, time-stamped audit trails that independently record the date and time of operator entries and actions that create, modify, or delete electronic records. Critically, the audit trail must capture previous and updated values — not just “a change occurred.”
- Retention and retrieval: Records must be protected and readily retrievable throughout the legally required retention period in both human-readable and electronic form.
- Access controls: System access must be limited to authorized individuals, with appropriate authority checks enforced at the application layer.
- Record integrity: Systems must use device checks and operational system checks to ensure that only authorized steps are performed in the proper sequence.
Electronic Signatures (§11.50, §11.100, §11.200): Each electronic signature must be:
- Unique to one individual — never reused or reassigned to another person
- Verified as truly belonging to that individual before first use
- Linked cryptographically or procedurally to the specific record it signs, preventing excision, copying, or transfer
- Accompanied by the signer’s printed name, the date and time of signing, and the meaning of the signature (approval, review, authorship, responsibility, etc.)
Non-biometric electronic signatures — i.e., username and password combinations — require at least two identification components, and credentials must be re-entered at the time of signing. Relying on an existing authenticated session is not sufficient.
Closed vs. Open Systems (§11.30): Open systems — those accessible to people outside the direct control of the organization — require additional controls, including encryption, digital signatures, and procedural controls to ensure record authenticity and integrity throughout the record’s lifecycle.
This sounds manageable on paper. The complexity lies entirely in implementation, validation, and ongoing lifecycle management — three disciplines that require explicit architectural planning from day one.
2. Common Misunderstandings: It Applies to More Systems Than You Think
The most dangerous assumption IT architects make is that Part 11 only applies to obvious “QA systems” — a dedicated LIMS, an electronic batch record platform, or a validation management tool. This assumption is wrong, and it carries serious regulatory consequences.
The Predicate Rule Concept
The FDA’s landmark 2003 guidance document on Part 11 scope introduced a concept that fundamentally reshapes how you must think about compliance: the predicate rule. A predicate rule is the underlying FDA regulation that requires a record to be created and maintained in the first place. Examples include:
- 21 CFR Part 211 — Current Good Manufacturing Practice for Finished Pharmaceuticals
- 21 CFR Part 820 — Quality System Regulation for medical devices
- 21 CFR Part 58 — Good Laboratory Practice for Nonclinical Laboratory Studies
- 21 CFR Parts 600–680 — Biological products regulations
- 21 CFR Part 312 — Investigational New Drug applications
Here is the key insight: Part 11 applies to any electronic record that is required by a predicate rule — or that is used to demonstrate compliance with a predicate rule — regardless of whether the FDA specifically mentioned that record type in Part 11 itself. The 2003 guidance also introduced the concept of enforcement discretion for certain lower-risk systems, but this is frequently misread as a blanket exemption. It is not.
Systems That Routinely Get Overlooked
Based on real-world audit findings and FDA warning letters, here are systems that organizations consistently underestimate as Part 11-relevant:
- HRMS and learning management systems — when used to manage training records for GMP-trained personnel
- Email and collaboration platforms (Teams, Slack, Outlook) — when used as the system of record for regulated approvals or decisions
- ERP systems (SAP, Oracle, Microsoft Dynamics) — when managing batch records, material traceability, supplier qualification, or change control data
- Data historians (OSIsoft PI, Ignition) — when capturing environmental monitoring data or process parameters required by predicate rules
- Cloud document storage (SharePoint, OneDrive, Google Drive, Box) — when used to store controlled SOPs, specifications, or batch records
- IT ticketing systems (ServiceNow, Jira) — when managing change control for validated systems
- Business intelligence platforms (Power BI, Tableau) — when generating compliance reports used in regulatory submissions
The FDA has issued warning letters citing Part 11 violations for systems that organizations never anticipated would receive scrutiny. The predicate rule concept means there is no safe assumption of exclusion. If the system creates or stores a record because a regulation requires it, Part 11 applies.
3. The 5 Things Auditors Always Check
FDA investigators conducting 21 CFR Part 11 inspections have a consistent playbook refined over more than two decades of audits. Understanding what they look for allows architects to design systems that pass — not just on paper, but under real investigator scrutiny.
1. Audit Trail Completeness
Auditors pull the audit trail for a specific record — a batch release, a deviation report, a calibration event, a CAPA — and trace every creation, modification, and deletion event from beginning to end. They look for gaps in the record, timestamp inconsistencies, and whether the trail captures who changed something, what it was before the change, what it became, and when (with full timestamp including timezone). Systems that only log “record modified” without capturing before/after values generate immediate Form 483 observations. So do systems that allow any user — including system administrators — to delete or alter audit trail entries.
2. Electronic Signature Authentication Strength
Auditors verify that electronic signatures cannot be executed by anyone other than the intended signer. At minimum, this means re-entry of username and password credentials at the time of signing — not a pre-existing session token or a “remember me” authenticated state. Investigators probe whether supervisors can sign on behalf of subordinates, whether service accounts can execute signatures, and whether multi-factor authentication (MFA) is in place for high-risk signature events. Industry best practices have shifted MFA from “recommended” toward functionally expected for any privileged signing action.
3. Access Control Logs
Who has access to the system? Why? When was access last reviewed? Auditors request full user access lists and compare them against active employees, contractors, and their documented roles. Finding an active account for a terminated employee — especially one with elevated privileges — is among the most common and most serious Part 11 findings. Investigators review role-based access matrices, compare authorized versus actual access, and ask specifically about privileged access and what compensating controls govern it.
4. Backup and Recovery Validation
Part 11 requires that electronic records be accurately and completely retrievable throughout the required retention period. Auditors don’t just ask “do you have backups?” — they ask “have you validated the restoration process?” They want documented backup validation protocols, execution records, restoration test results, and evidence that restored records are intact and unaltered. Backup frequency, retention periods, off-site or off-region storage, and encryption of backup media are all within scope of the inspection.
5. Change Control Records
Any change to a validated system — a software update, configuration modification, infrastructure change, or vendor-initiated patch — requires a change control record documenting the nature of the change, risk assessment, validation activities performed, and approvals obtained. Investigators regularly find that SaaS vendor updates or cloud platform changes proceeded without any client-side change control or validation impact assessment. This is among the most systemic vulnerabilities for organizations using hosted and cloud-based regulated applications.
4. Link to GAMP5: The Risk-Based Approach
One of the most practically valuable frameworks for Part 11-compliant system architecture is GAMP5: A Risk-Based Approach to Compliant GxP Computerized Systems, published by the International Society for Pharmaceutical Engineering (ISPE). Now in its second edition (2022), GAMP5 provides the pharmaceutical and life sciences industry’s most widely accepted methodology for categorizing systems by risk and applying proportionate validation effort.
GAMP5 Software Categories
GAMP5 classifies software into four categories that guide validation depth and documentation requirements:
- Category 1 — Infrastructure Software: Operating systems, database engines, middleware, and network software. Focus on platform qualification rather than full application validation.
- Category 3 — Non-Configured Software: Standard, packaged software used as-is (e.g., standard laboratory instruments with embedded firmware, off-the-shelf tools with no configuration).
- Category 4 — Configured Software: Commercial off-the-shelf (COTS) applications with configuration but no custom code — ERP, LIMS, MES, DMS, CTMS. This category represents the primary focus of most Part 11 validation efforts in life sciences organizations.
- Category 5 — Custom Software: Bespoke applications or systems containing custom-developed code. Highest validation burden, requiring full software development lifecycle documentation including design specifications, code review, and unit testing evidence.
How GAMP5 Guides Part 11 Implementation
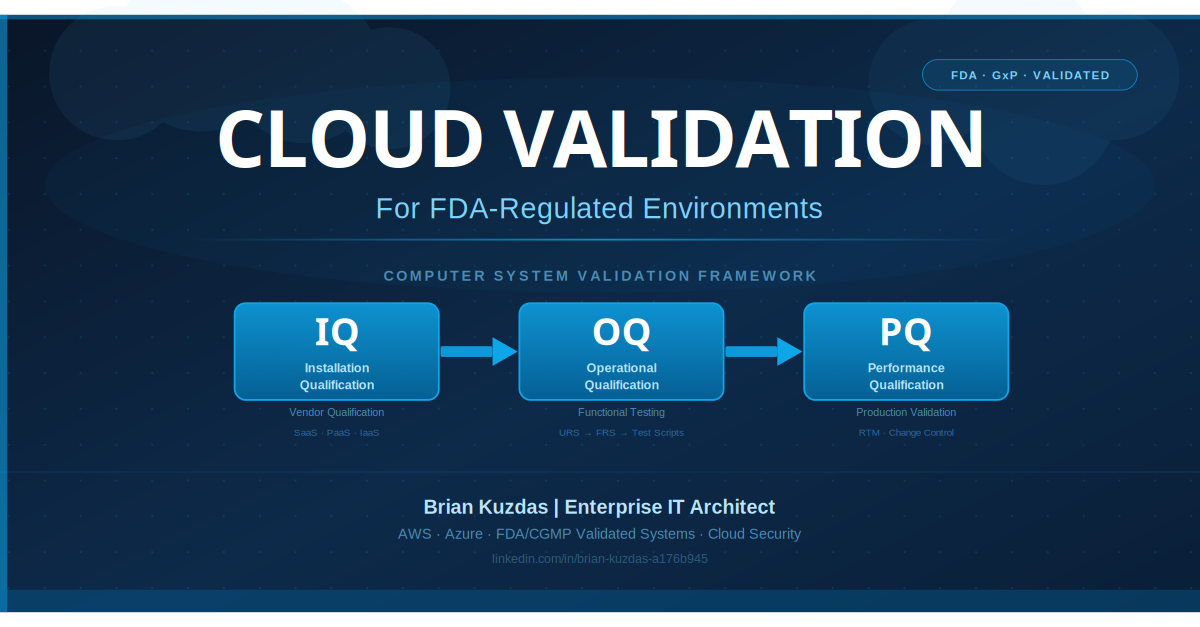
GAMP5’s risk-based approach means validation effort is proportionate to risk — not uniform across all systems. A Category 4 LIMS managing batch release records receives a full Validation Life Cycle: User Requirements Specification (URS), Functional Requirements Specification (FRS), Design Qualification (DQ), Installation Qualification (IQ), Operational Qualification (OQ), and Performance Qualification (PQ), along with a Validation Summary Report. A Category 1 database server supporting that LIMS receives platform qualification documentation without the full application validation lifecycle.
Key GAMP5 principles for architects to internalize:
- Intended use determines risk level — not the underlying technology or cost of the system
- Validation is a lifecycle activity, not a one-time event; every change triggers a validation impact assessment
- Supplier assessment reduces (but does not eliminate) the validation burden for Category 4 systems; the regulated company retains compliance responsibility
- All risk decisions must be documented and defensible — the rationale for each validation decision must withstand FDA investigator review
GAMP5 also aligns closely with EU GMP Annex 11: Computerised Systems, the European equivalent regulatory framework. Organizations with global regulatory obligations — submitting to both FDA and EMA — will find that designing systems to satisfy both frameworks from the outset is far more efficient than retrofitting for either after initial deployment.
5. How Cloud Complicates Things: The Shared Responsibility Gap
The migration of life sciences workloads to cloud platforms — AWS, Azure, Google Cloud — and the proliferation of SaaS applications creates a fundamental tension with 21 CFR Part 11: the regulated entity remains fully responsible for compliance, regardless of how much infrastructure is managed by a third party.
The Shared Responsibility Model Is Not a Compliance Defense
Cloud providers’ shared responsibility models define who secures which layer of the technology stack: the provider secures the physical data centers, hardware, and hypervisor layer; the customer secures what they deploy on top. This is a cybersecurity and operational model. The FDA does not recognize it as a regulatory compliance framework. If an underlying cloud platform change — a hypervisor update, storage system modification, or networking infrastructure change — causes a Part 11 compliance gap, the regulated company bears the full regulatory consequence.
SaaS Platform Challenges
SaaS-hosted regulated applications present unique and layered compliance challenges:
- Limited visibility: The customer typically cannot inspect how the SaaS vendor implements audit trails, access controls, or encryption at the infrastructure level. What you see in the application UI may not reflect everything — or enough — about the underlying implementation.
- Vendor update cadence: SaaS vendors push updates on their own schedule, often with minimal advance notice. An update that modifies data handling, workflow logic, or audit logging behavior may require the client to perform a change control assessment and validation impact analysis — even if the client had no input into the change.
- Data residency: Records must be retained in accessible, controllable storage that meets Part 11 requirements. Organizations must contractually verify where SaaS vendor data actually resides, including backup copies and disaster recovery sites.
- Supplier qualification: Every SaaS vendor used for a Part 11-relevant system must be formally qualified. This requires audit rights, review of SOC 2 Type II reports, evaluation of the vendor’s validation documentation, and contractual commitments for advance change notification.
IaaS and PaaS Validation Architecture Patterns
For infrastructure-as-a-service (IaaS) and platform-as-a-service (PaaS) deployments, the validation burden sits primarily with the regulated organization. Practical architecture patterns that support Part 11 compliance in cloud environments:
- Infrastructure-as-Code (IaC): Use Terraform, Pulumi, or CloudFormation with version-controlled templates deployed through change-controlled CI/CD pipelines. This creates reproducible, documented system configurations that can serve as the basis for IQ/OQ documentation.
- Centralized compliance logging: Feed AWS CloudTrail, Azure Monitor, or Google Cloud Audit Logs into a tamper-resistant SIEM or dedicated compliance logging platform. Logs must be protected from modification and retained for the legally required period.
- Cloud key management: Use AWS KMS, Azure Key Vault, or Google Cloud KMS for encryption key management with full key usage audit trails maintained independently of application data.
- Privileged access management: Separate system administrative access from normal production access. Implement just-in-time (JIT) privileged access with full logging and session recording.
- Automated backup validation: Automate restoration tests on a scheduled basis and generate machine-readable evidence records as part of your validated backup and recovery process.
6. Practical Implementation Checklist
Whether you are architecting a new regulated system from the ground up or assessing an inherited implementation with uncertain compliance status, this checklist provides a structured starting framework.
System Scoping and Classification
- ☐ Identify all electronic records created or maintained by the system
- ☐ Determine the predicate rule(s) that require these records
- ☐ Classify the system using GAMP5 categories (1, 3, 4, or 5)
- ☐ Document the intended use and complete a formal risk assessment
- ☐ Determine whether the system is closed (§11.10) or open (§11.30)
Access Controls
- ☐ Implement role-based access control (RBAC) with documented role-permission matrices
- ☐ Ensure all system accounts are individual — no shared accounts permitted
- ☐ Establish a formal user access review process (at minimum annually)
- ☐ Integrate account deprovisioning with HR offboarding workflows
- ☐ Confirm terminated employee accounts are disabled within 24 hours of separation
- ☐ Document and control all privileged/administrative access separately
Audit Trails
- ☐ Verify audit trail captures: user ID, timestamp with timezone, action type, record ID, previous value, new value
- ☐ Confirm audit trail is computer-generated and cannot be modified or deleted by any user, including administrators
- ☐ Test audit trail completeness across all create, modify, and delete operations
- ☐ Include periodic audit trail review in system review procedures
- ☐ Protect audit trail storage with access controls and integrity verification
Electronic Signatures
- ☐ Verify signatures require re-entry of authentication credentials at the time of signing
- ☐ Confirm signature records contain: signer name, date/time, signature meaning
- ☐ Verify signatures are cryptographically or procedurally linked to their respective records
- ☐ Confirm signature delegation and shared signing are technically prevented
- ☐ Document the electronic signature procedure in a formal SOP
Validation Documentation
- ☐ Produce a User Requirements Specification (URS) capturing all regulated requirements
- ☐ Develop and execute IQ (Installation Qualification) protocol
- ☐ Develop and execute OQ (Operational Qualification) with documented test scripts and results
- ☐ Produce a Validation Summary Report reviewed and approved by QA
- ☐ Establish change control procedures for all ongoing system modifications
Backup and Recovery
- ☐ Document backup frequency, retention period, and storage location
- ☐ Develop and execute a backup restoration test protocol
- ☐ Store restoration test results as validated, Part 11-compliant records
- ☐ Review backup and recovery procedures at minimum annually
Supplier and Cloud Vendor Management
- ☐ Conduct formal supplier qualification assessment for each cloud/SaaS vendor
- ☐ Obtain and review vendor SOC 2 Type II reports (annual)
- ☐ Establish audit rights in vendor contracts
- ☐ Review vendor’s own validation documentation where available
- ☐ Define vendor change notification requirements contractually
- ☐ Assess and document data residency, retention, and backup capabilities
The Bottom Line
FDA 21 CFR Part 11 compliance is not a checkbox activity. It is an architectural discipline that must be designed in from the beginning, maintained through the full system lifecycle, and documented rigorously enough to survive the scrutiny of an FDA investigator who has reviewed thousands of implementations — successful and failed alike.
The cost math is unambiguous. A Form 483 observation requires a formal written response within 15 business days and can trigger follow-up inspections, consent decrees, import alerts, or criminal referrals in the most serious cases. Industry experience consistently places the total cost of responding to a Form 483 — accounting for internal resource hours, compliance consultants, corrective and preventive actions (CAPAs), remediation work, and potential impact on product availability — at $500,000 to well over $2 million per observation. FDA Warning Letters, the formal escalation above a Form 483, carry stock price impacts, import bans, and reputational consequences that dwarf any upfront validation investment.
The FDA warning letters database (FDA.gov) is publicly searchable. Reviewing it for 21 CFR Part 11 and computerized system violations reveals a consistent, predictable pattern of failures: missing or incomplete audit trails, inadequate electronic signature controls, unvalidated or improperly validated systems, and failure to maintain change control records for regulated systems. These are not exotic edge cases. They are the predictable outcomes of underinvesting in validation architecture at the design stage.
Practical takeaways for IT architects:
- Ask the predicate rule question for every system that touches regulated processes. If the system creates or maintains a record that any FDA regulation requires, Part 11 applies — regardless of how peripheral the system appears.
- Design audit trails first. Retrofitting audit trail functionality into an existing system is one of the most expensive and disruptive activities in validated system management. Build it in from day one.
- Validation is a lifecycle, not an event. Every change — including vendor SaaS updates you didn’t request — requires a documented impact assessment and, where appropriate, revalidation activities.
- Cloud shared responsibility is a security model, not a compliance model. You own Part 11 compliance regardless of where the system runs or who manages the underlying infrastructure.
- Use GAMP5 to right-size your effort. Risk-based validation means applying rigorous validation to high-risk, high-impact systems and proportionate effort to lower-risk infrastructure. Document the rationale. Make every decision defensible.
The organizations that genuinely succeed with Part 11 don’t treat it as a regulatory burden imposed from outside. They treat it as an engineering quality standard — and in FDA-regulated industries, that distinction is what separates companies that grow from companies that face consent decrees.
References
-
U.S. Food & Drug Administration. 21 CFR Part 11 — Electronic Records; Electronic Signatures (Title 21, Code of Federal Regulations, Parts 11.1–11.300). https://www.ecfr.gov/current/title-21/chapter-I/subchapter-A/part-11
-
U.S. Food & Drug Administration. Guidance for Industry: Part 11, Electronic Records; Electronic Signatures — Scope and Application (August 2003). https://www.fda.gov/media/75414/download
-
International Society for Pharmaceutical Engineering (ISPE). GAMP5: A Risk-Based Approach to Compliant GxP Computerized Systems, Second Edition (2022). https://ispe.org/publications/guidance-documents/gamp-5-guide-2nd-edition
-
European Commission. EudraLex Volume 4: EU Guidelines for Good Manufacturing Practice — Annex 11: Computerised Systems (January 2011). https://health.ec.europa.eu/system/files/2016-11/annex11_01-2011_en_0.pdf
-
U.S. Food & Drug Administration. Warning Letters Database. https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/compliance-actions-and-activities/warning-letters
-
ISPE. GAMP Good Practice Guides (series, various dates). https://ispe.org/publications/guidance-documents
-
U.S. Food & Drug Administration. Cybersecurity in Medical Devices: Quality System Considerations and Content of Premarket Submissions (September 2023). https://www.fda.gov/regulatory-information/search-fda-guidance-documents/cybersecurity-medical-devices-quality-system-considerations-and-content-premarket-submissions
-
U.S. Food & Drug Administration. 21 CFR Part 211 — Current Good Manufacturing Practice for Finished Pharmaceuticals. https://www.ecfr.gov/current/title-21/chapter-I/subchapter-C/part-211
#FDA #21CFRPart11 #CGMP #Compliance #ITArchitecture #Validation #LifeSciences #CloudCompliance
Brian Kuzdas is an Enterprise IT Architect based in Madison, WI with 25+ years of experience in enterprise IT. He specializes in FDA/CGMP validated systems, cloud security, zero trust architecture, HL7/FHIR, and Identity and Access Management (IAM). He holds certifications from AWS, Azure, Okta, Databricks, and Google Cloud. Connect on LinkedIn: https://linkedin.com/in/brian-kuzdas-a176b945